Hypertrophic cardiomyopathy: Do you know what to do if your heart is at risk?
)
While the risk of sudden death has for a long time been emphasised, more balanced perspectives on hypertrophic cardiomyopathy are emerging.
Hypertrophic cardiomyopathy (HCM) is characterised by a thickening of the heart muscle in the ventricular wall, usually the left ventricle. The overall ventricle size often remains normal but the inside of the ventricle is generally smaller and holds less blood than a normal ventricle, impeding normal cardiac function. The American Heart Association claims that HCM is relatively common, occurring in 1 out of every 500 people, and is the leading cause of sudden cardiac arrest in young people (less than 30 years of age), including young athletes, in the United States (US). This has stimulated calls for the routine HCM screening of athletes.
Hypertrophic cardiomyopathy (HCM) is a complex and often contentious area within cardiology, particularly concerning its diagnosis and management. The controversies in diagnosing and managing HCM stem from its varied presentation, the genetic underpinnings, and the balance between managing symptoms and preventing complications.
While the adverse consequences of HCM (i.e.the risk of sudden death) have been emphasised in the literature, more balanced perspectives on HCM are emerging. The overall mortality rates for HCM in the US (around 1% per year) are, in fact, not dissimilar to the general US adult population. Many people with HCM go undiagnosed because they never have signs or symptoms and they can actually lead normal lives without significant problems. Others may experience classic signs and symptoms (e.g., shortness of breath, fainting, irregular heart rhythms) leading to diagnosis and appropriate management.
Diagnostic challenges
The diagnosis of HCM can be fraught with difficulties due to its heterogeneous nature. The primary diagnostic tool is echocardiography (cardiac ultrasound), which reveals the characteristic thickened ventricular walls. However, distinguishing HCM from other conditions that cause left ventricular hypertrophy, such as athletic heart syndrome or hypertension, can be challenging. Genetic testing has emerged as a valuable tool, identifying mutations in genes encoding sarcomeric proteins. Despite this advancement, the interpretation of genetic variants can be complex, with many variants of uncertain significance (VUS), which complicates the diagnosis.
Another layer of controversy involves the differentiation between HCM and other forms of hypertrophic remodeling. For instance, distinguishing HCM from hypertensive heart disease can be particularly challenging in older patients who may have both conditions. Moreover, the advent of advanced imaging techniques, such as cardiac magnetic resonance imaging (MRI), has improved the accuracy of HCM diagnosis but has also highlighted the complexity of phenotypic expression, with some patients exhibiting minimal or atypical hypertrophy.
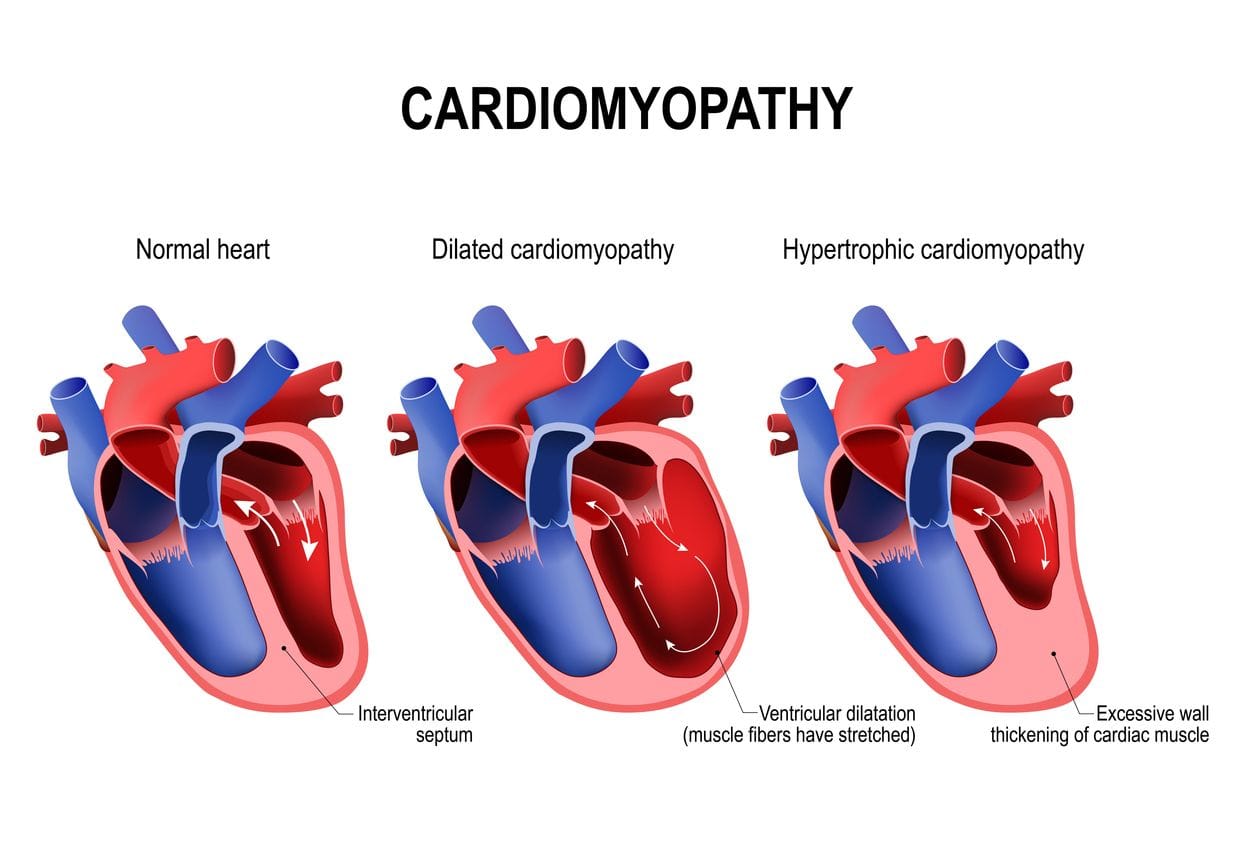
Management controversies
Once diagnosed, the management of HCM presents another set of controversies. The primary goals of management are to alleviate symptoms, prevent complications, and reduce the risk of sudden cardiac death. However, achieving these goals is not straightforward and often requires a tailored approach.
Risk stratification and prevention of sudden cardiac death
One of the most debated aspects is risk stratification for sudden cardiac death. The implantation of an implantable cardioverter-defibrillator (ICD) is a proven method to prevent sudden death in high-risk patients. However, identifying those at highest risk remains contentious. Traditional risk factors include a family history of sudden death, unexplained syncope, severe left ventricular hypertrophy, and non-sustained ventricular tachycardia. Recent guidelines, such as those of the European Society of Cardiology, also incorporate a risk calculator that considers multiple factors to estimate an individual's risk. Despite these tools, predicting sudden cardiac death is not foolproof, and the decision to implant an ICD can be challenging, particularly in younger patients due to the potential for device-related complications over a lifetime.
Pharmacological and non-pharmacological treatment
The primary objectives of pharmacological treatment in HCM include the relief of symptoms, the management of left ventricular systolic dysfunction and heart failure (which occur when the left side of the heart does not pump efficiently or contract the way it should between heartbeats), the management of arrhythmias (irregular heartbeats), and the prevention of cardio-embolic events (strokes). Pharmacological management primarily involves beta-blockers, calcium channel blockers, and disopyramide to relieve symptoms and improve exercise tolerance. However, the response to these medications can be variable, and side effects are not uncommon. Septal reduction therapy, either through surgical myectomy or alcohol septal ablation, is another option for patients with obstructive HCM who do not respond to medical therapy. These procedures can significantly alleviate symptoms but carry their own risks and are subjects of debate regarding their long-term outcomes and optimal patient selection.
Lifestyle and activity recommendations
The recommendations regarding physical activity for HCM patients are another area of controversy. While moderate-intensity exercise is generally encouraged for overall cardiovascular health, high-intensity and competitive sports are typically discouraged due to the increased risk of adverse cardiac events. However, recent studies suggest that some patients with HCM may safely participate in certain levels of exercise, prompting a re-evaluation of existing guidelines and emphasizing the need for individualised exercise prescriptions.
Conclusion
The diagnosis and management of hypertrophic cardiomyopathy remain complex and contentious due to the disease's heterogeneous nature and the challenges in balancing symptom relief with the prevention of complications. Ongoing research and advances in genetic testing, imaging, and risk stratification tools continue to evolve the landscape, but the need for individualized patient care remains paramount. Clinicians must navigate these controversies with a nuanced understanding of the disease and a commitment to personalised treatment strategies.
--
References
Elena Arbelo E et al. (2023). 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC), European Heart Journal, 44(37): 3503-3626. https://doi.org/10.1093/eurheartj/ehad194
Corliss J (2023). Advances in managing hypertrophic cardiomyopathy. Harvard Health Publishing, Harvard Medical School. https://www.health.harvard.edu/heart-health/advances-in-managing-hypertrophic-cardiomyopathy
Monda E, Limongelli G, Pelliccia F. (2023). Hypertrophic cardiomyopathy - Current challenges and future perspectives. Journal of Clinical Medicine, 12(18):6093. doi: 10.3390/jcm12186093. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10531662/
Semsarian, C, Gray, B, Haugaa, K. et al. (2022). Athletic activity for patients with hypertrophic cardiomyopathy and other inherited cardiovascular diseases: JACC Focus Seminar 3/4. Journal of the American College of Cardiology, 80(13): 1268-1283.https://doi.org/10.1016/j.jacc.2022.07.013
Zhang Y et al. (2024). Management of hypertrophic cardiomyopathy. Journal of Cardiovascular Medicine, 25(6):399-419. doi: 10.2459/JCM.0000000000001616. Epub 2024 Apr 26. PMID: 38625835; PMCID: PMC11142653. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11142653/
)
| Tags:Heart problemsHeart Health for AthletesHH4A |